HEAL Committee Report
If you have any questions or comments regarding the accessibility of this publication, please contact us at accessible@parl.gc.ca.
- CHAPTER 4 - EXPERTISE AND REGULATORY STRUCTURE
- OPTIONS FOR THE NEW REGULATORY AUTHORITY
- CHAPTER 5 - REGULATORY FRAMEWORK
- CHAPTER 6 - LABELLING
- CHAPTER 7 - SECTION 3 AND SCHEDULE A OF THE FOOD AND DRUGS ACT
PART II: REGULATION
AND REGULATORY STRUCTURE
Many witnesses asserted that the existing level of expertise and experience within Health Canada was insufficient to review NHPs for approval for sale on the Canadian market or to ensure appropriate post-market surveillance. They explained that, although employees of the department may have attempted to learn about NHPs, they do not fully understand either how these products work or the philosophical and cultural background behind them. A number of witnesses even suggested that some personnel at the department were openly hostile toward natural health and that they exercised their authority in an overtly partisan way.
Currently, there are no naturopaths, herbalists, homeopaths or traditional Chinese, North American and Ayurvedic representatives working within Health Canada or the Canadian Food Inspection Agency. Witnesses claimed that this situation has led to a regulatory environment fraught with confusion. Policies and regulations have been and continue to be developed by non-experts. Products are removed from the market or moved from food to drug status, often without a rational justification by anyone who has experience in NHPs. Field inspectors, including those working for Customs, the Canadian Food Inspection Agency as well as the Health Protection Branch, frequently do not have an adequate knowledge of NHPs and, as a result, do not uniformly enforce regulations across the country.
The Committee was also told that the HPB closed its sole natural products division Dr. Dennis Awang headed this division, which was within the chemistry division of the Bureau of Drug Research. The natural products division was involved in the investigation of particular hazards relating to NHPs. According to documents provided to the Committee, this NHP section was well established and internationally recognized. However, it was dismantled with the closing of the Bureau of Drug Research in 1991. Recently, a Natural Health Product Division was established within the Drug Assessment Bureau of the TPP. This new division, however, does not perform the same functions as the former NHP division, nor does it have the capabilities to properly assess NHPs for the purpose of sale.
Therefore, witnesses urged that the administration of the regulatory framework pertaining to NHPs be guided by the expertise of individuals skilled in the field of natural health and, accordingly, they recommended that a new regulatory authority or structure be established. There was confusion, however, as to the precise structure and organization for this new entity. The APNHP did not reach a unanimous consensus on this issue.
The Committee strongly agrees that decisions concerning NHPs must be made by people with expertise and understanding of the products. Committee members also acknowledge that decisions by the new regulatory structure must not be overturned by others without expertise. For these reasons, the Committee considered four major options which were drawn from the testimony. These options are illustrated in Table 1.
TABLE 1
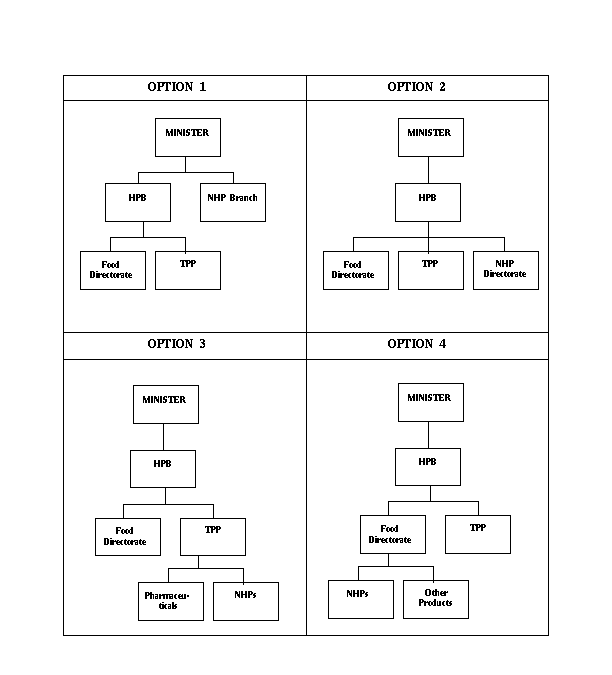
Option 1 proposes that the new regulatory authority be within Health Canada but separate from the HPB. This option was recommended by those in favour of a separate category for NHPs (some witnesses even suggested that an NHP agency be created outside Health Canada). In Option 2, the regulatory authority responsible for NHPs would be separate from, and independent of, both the Food Directorate and the TPP, but would still fall under the HPB. This option was suggested by the advocates of a new process for these products, distinct from both foods and drugs, but with regulations administered by the HPB. Option 3 assumes that responsibility for NHPs would lie within the TPP but that NHP regulations would be different from that of pharmaceutical products. This option was favoured by those who suggested that, while NHPs are different from pharmaceutical products, the pre-market assessment and post-market surveillance for both categories of products share similar characteristics. Finally, Option 4 assumes that the Food Directorate would be in charge of administering NHP regulations. This option was mainly recommended by witnesses representing consumers who contended that the current food regulations are adequate for NHPs. They stated that no new regulations were necessary and that what was needed was the effective enforcement of sections 4 and 5 of the Food and Drugs Act.
The Committee considered these 4 options very seriously. All four options are consistent with the guiding principles relating to decision-making and the nature of NHPs. However, both Options 3 and 4 are problematic for the Committee. As mentioned earlier, witnesses argued that the existing levels of expertise and experience within both the Food Directorate and the TPP are insufficient to address NHPs. Some linked this lack of experience and expertise to a negative organizational bias against NHPs. The Committee in rejecting Option 3 and Option 4 gave serious consideration to these views. However, its final assessment is based on its own belief that the assessment of NHPs would benefit from a fresh approach, one where NHPs are evaluated in a forum and through a process detached from either pharmaceuticals or foods.
Options 1 and 2 come closer to satisfying those who requested an administrative body separate from existing regulatory structures and modes of operating within the HPB. Both would ensure more independent decision-making with respect to NHPs. However, over the course of this study, the Committee has become aware of the level of information, equipment and personnel needed to support regulatory activities aimed at ensuring safety for Canadians. Still, the need for a new entity with a separate, small body of permanent staff is supportable. This can best be achieved without replication of existing bureaucracy and without significant cost if the new regulatory authority is placed within the HPB where it can have proximity and access to existing regulatory resources. Thus, the Committee sees Option 2 as more likely than Option 1 to result in low administrative costs. In addition, placing the NHP authority within the HPB as a directorate on equivalent terms with foods and pharmaceuticals reinforces the Committee's view that NHPs are different from, but complementary to, these other products for health.
The structure envisioned by the Committee reports directly to the Assistant Deputy Minister of the Health Protection Branch. With reference to lower administrative costs, the Committee members see it as consisting of a small number of permanent, full-time staff drawing external support as needed from the newly-created NHP Expert Advisory Committee and appropriate working groups. We feel that there can be a sharing of relevant resources already existing within HPB and a greater use of electronic linkages to facilitate information flow from outside resources and experts across Canada.
Committee members also want to stress that time is of the essence. The Committee agrees with the overwhelming view offered by witnesses that no one should tolerate a long wait for the system to be modified, which could happen if we have to wait for legislative changes. The Committee feels that appropriate regulatory and administrative changes can and should be affected, with the necessary legislative modifications to follow. The new regulatory authority can and should be established as soon as possible: a six-month period seems quite reasonable.
It may be difficult to establish a new structure with staff from all the various fields of NHPs. For this reason, the regulatory authority for NHPs, which would include only a limited number of administrators, should have the ability to establish working groups when necessary to examine specific products, specific claims, etc. These working groups would report their findings to the new regulatory authority. The Committee believes that in-house expertise, combined with relevant external consultation through electronic networking, will result in appropriate management of the NHP regulatory framework.
At this time, the Committee feels that it may not be necessary to hire new inspection staff. However, all inspectors dealing with NHPs must be provided with training specific to NHPs.
Overall, the Committee recommends that:
The Government give consideration to the advisability of creating a new regulatory authority for NHPs that reports directly to the Assistant Deputy Minister of the Health Protection Branch;
The structure for this new regulatory authority be established within the next six months and be permanently staffed by individuals with expertise and experience in the field of NHPs;
The selection of personnel be agreeable to both government and NHP stakeholders;
When necessary, working groups reflecting the various segments that make up the NHP category be set up to advise the new regulatory authority;
All relevant inspection personnel be provided with training specific to NHPs;
The necessary process to amend the Food and Drugs Act not delay in any way the implementation of the regulatory and administrative changes that can proceed at this time.
The Committee agrees with many witnesses that, in creating the new regulatory environment, a new, external, independent Expert Advisory Committee must be established immediately. Its general tasks will include assistance to Health Canada in developing new regulations, revising legislation, and setting out appropriate policies. Some of the more specific work such as defining NHPs, developing safety protocols, etc. is indicated in other Committee recommendations. This Expert Advisory Committee should also examine the closure of the former NHP section headed by Dr. Awang with a view to proposing that it be re-established or that a different laboratory entity be created. Members of the Expert Advisory Committee should be required to provide full-time commitment until all their tasks are fulfilled. In addition, they should be reconvened at least once a year, or more frequently if necessary, to report on the progress of the new regulatory framework. The Expert Advisory Committee should report to the new regulatory authority and its findings should be made public. In line with the Committee's guiding principle, this would ensure the transparency of the overall regulatory system. Decisions regarding NHPs must be made in conjunction with those who have both the expertise and an understanding of the products and how they are used by consumers or recommended by practitioners.
Therefore, the Committee recommends that:
An Expert Advisory Committee be established immediately to assist Health Canada in the general and specific tasks necessary to design a new NHP regulatory environment;
This Expert Advisory Committee review the re-establishment options for an NHP section with research and laboratory capacities and report its findings to Health Canada;
The selection of members for the Expert Advisory Committee be agreeable to both NHP stakeholders and Health Canada.
For Committee members, the subject of product assessment in relation to NHPs focused primarily on three areas: safety, quality and efficacy. Witnesses repeatedly raised questions in this regard. On safety, they asked: Will the product cause harm if taken as suggested? What are its side effects, if any? Are there any risks associated with its use? On quality, questions included: Is it what it says it is? What guarantees that the product is as described on the label? In relation to efficacy, the questions were: Will the product work as claimed? Will it improve health outcomes?
From meetings with both Health Canada and the Canadian Food Inspection Agency, members heard that these issues were addressed through assessments at various stages of production for foods and drugs. According to representatives from both bodies, their assessments are based on measures aimed at identifying the appropriate balance between possible threats and/or potential benefits to human health if the product is consumed.
The task of reconciling divergent opinions about the appropriate risk/benefit ratio to apply to individual NHPs was a major one for Committee members. We became aware that, in assessing products for human consumption, the terms "risk" and "benefit" have particular meanings. Thus, risk is generally defined as the probability of the occurrence of an adverse event from exposure to a substance combined with the harm to human health if such an event occurs. Benefit is defined as the human health improvement attributable to the product. Risk analysis includes identifying the existence of a hazard and estimating the probability of its occurrence; benefit analysis involves recognizing the existence of a benefit and measuring the kind and degree of improvement. Traditionally, where risks have been related to safety, the benefits of a product have been linked to its efficacy or desired results under ideal or recommended conditions. Thus, while risk might be measured in terms of increases in morbidity and mortality, benefits could be measured by reductions in these.
The assessment of NHPs using this accepted risk/benefit model poses particular problems. Both the risks and the benefits associated with use of NHPs are perceived differently by the various involved groups. For example, regulators who see protection of consumers as a paramount responsibility focused on documented evidence of adverse effects while members of the public who are informed consumers practising self-care emphasized their experience of positive health benefits. This situation was further complicated by different assessments emanating from those who viewed NHPs as foods, those who identified them as drugs, and those who saw them as different from either of these. Emerging from the ongoing discussion was a strong sense that the current emphasis on level of risk misrepresents the majority of NHPs and that it would be more useful to approach the products from a perspective stressing margins of safety.
In order to understand how this type of analysis might apply to NHPs, the Committee members sought greater understanding of its current applications for foods and for drugs. We wanted more knowledge of the ongoing assessment process of products intended for human consumption. This process which begins before a product is marketed continues after it is available for sale. In the case of foods and of drugs, assessment can begin at a laboratory, or in the case of plants at a greenhouse and follow through to the post-consumption stage. Once production begins, good manufacturing practices, whether mandated through regulation or voluntary through guidelines, are crucial to ensuring safety and quality, if not also efficacy.
2. Pre-market Product Assessment
Although the Food and Drugs Act prohibits the sale of any food or drug that has been adulterated or represented in a manner that is false, misleading or deceptive, the pre-market approval process for foods is very different from that for drugs.
NHPs sold as foods are generally not subject to pre-market evaluation and approval requirements. As with most other food products, it is the responsibility of manufacturers/sellers/importers to ensure that NHPs are safe. In this regard, Section 4 of the Act provides as follows:
No person shall sell an article of food that (a) has in or on it any poisonous or harmful substance; (b) is unfit for human consumption; (c) consists in whole or in part of filthy, putrid, disgusting, rotten, decomposed or diseased animal or vegetable substance; (d) is adulterated; or (e) was manufactured, prepared, preserved, packaged or stored under unsanitary conditions.
With safety in mind, Health Canada can prohibit products from being sold as foods due to their inherent harmfulness. At the beginning of the Committee's study, there was a list of sixteen NHPs deemed to be unacceptable as foods or components of foods. This list was drawn from Schedule 705 which proposed amendments to the Food and Drug Regulations pursuant to an earlier assessment of herbs and botanical preparations. The Committee was told that the assessment of potential risks from various hazards encountered in food production systems is usually based on toxicological and epidemiological data. Despite the absence of pre-market approval requirements, Health Canada's Food Directorate does use available data to provide potential sellers with opinions on the safety of various herbal preparations and components. In addition, pre-clearance of the safety of all "additives" is required, as is the establishment of maximum residue limits for pesticides in foods.
The Committee heard that, when NHPs are sold as foods, they constitute an exceedingly small proportion of the total Canadian food system and are also considered to represent a low health or safety risk relative to other staple foods. As such, both Health Canada's Food Directorate and the Canadian Food Inspection Agency (CFIA) assign a lower priority to assessments and scrutiny of this group of products. These government bodies conduct assessments of potential problems on a case-by-case, product-by-product basis. Thus, if it is determined that any NHP sold as a food poses a threat to consumer health, the Food Directorate of Health Canada will recommend to the CFIA that appropriate compliance action be taken.
By contrast, all products sold as drugs, including NHPs, must be assessed and pre-approved before they can be marketed. Generally speaking, there are different ways of evaluating new products, known products for new uses, prescription products and non-prescription products. The pre-market evaluation is based on an analysis of benefits and risks associated with each drug product: the higher the risk, the greater the level of evidence and data required. When an evaluation shows a positive benefit-risk ratio, a market authorisation is issued by means of a Drug Identification Number (DIN) or a General Public Number (GP). This number indicates that a product has successfully passed through a review of its formulation and labelling.
The TPP has developed and implemented particular processes for each of the three categories of NHPs included in our Committee's mandate - herbal products, homeopathic preparations, and vitamin and mineral supplements.
For traditional herbal medicines, submissions to obtain DINs must comply with the related guidelines and policy. Firstly, traditional herbal medicines must present no safety concerns. Secondly, each submission must include traditional herbal references, a monograph, reputed pharmacological actions, dosage and other information. Lastly, the indications for use must be consistent with the principles of self-medication: consumers must be able to understand clearly the purpose of products. Approval of traditional herbal medicines is based mainly on traditional herbal references, provided that the herbal medicines are not known to be unsafe in treating minor ailments. Those herbal products not satisfying traditional herbal medicine criteria are assessed through a different process. Manufacturers wishing to market herbal medicines for the treatment of more serious ailments must provide supporting scientific and clinical data; at present, there are few herbal medicines with such data.
Homeopathic preparations and indications for their use must also be approved. Firstly, indications for use must be suitable for self-diagnosis and self-treatment and correspond to self-limiting conditions. Secondly, indications for use are permitted on the labels of multi-ingredient low-dilution homeopathic preparations, but not single-ingredient or multi-ingredient intermediate- or high-dilution homeopathic preparations. Lastly, a Health Canada Labelling Standard applies if a manufacturer does not wish to include indications for use on a label. In this case, the medicinal ingredients and their concentrations are restricted to those specified in the Labelling Standard. Having recognised that sections C.01.036, C.01.038 and C.01.040 of the current Regulations block legal access to many commonly used, high-dilution, low-risk homeopathic preparations manufactured from certain potentially toxic prohibited substances, the TPP has made recommendations to revoke these sections. This change would give Health Canada the flexibility to evaluate homeopathic preparations manufactured from prohibited substances on the basis of their benefits and risks. According to Health Canada, this change would provide legal access to these products, without compromising consumer safety where higher-risk products are concerned.
Vitamins and mineral supplements are generally classified as products that present minimal risks. Extensive prior knowledge and experience regarding the safety, efficacy and quality of their active ingredients are widely available. Health claims and daily limits for vitamins and minerals are regulated under Divisions 4 and 5 of Part D of the Regulations. Some observers consider these Regulations restrictive: Health Canada requires manufacturers wishing to include health or therapeutic claims beyond the scope of claims contained in the Regulations to provide objective scientific evidence in support of these expanded claims. The department is considering the possibility of amending the Regulations in order to expand allowable health claims for vitamin and mineral supplements.
3. Post-market Product Assessment
Post-market surveillance continues the monitoring of risks and benefits. The level of monitoring after marketing is based on the degree of risk attributed to the product. The monitoring can take place during production and after consumption.
For drugs, this monitoring is required through regulations that outline procedures for reporting side-effects and for GMP licence renewals. Thus, regulations require the collection and analysis of post-approval adverse drug reaction data. When authorized to market, the drug must be produced in adherence with GMP standards that seek to ensure that quality and safety are ensured.
For foods, equivalent monitoring activities are not regulated. The collection of data on adverse reaction to herbs and botanicals sold as foods is not required and no system is currently in place. Production practices and facility standards are not regulated; instead voluntary compliance with recommended guidelines aimed at safety and quality and developed through consensus by government and industry is encouraged.
4. Relevant Proposals for Drugs and Foods
Ongoing consultations by the federal government have yielded suggestions for changes to the way both drugs and foods and "in-between products" might be assessed.
The ongoing work to develop an appropriate regulatory framework for products being called nutraceuticals and functional foods has relevance for NHPs. As noted earlier, the working definitions developed for both categories of products have applicability to various products brought to the attention of the Committee. Thus, current ways of distinguishing between foods and drugs must be re-evaluated to include products seen as having important nutritional elements with the potential to prevent disease or to modify physiological functions.
More specifically, related to drugs, the gradual implementation of the Product Licensing Framework (PLF) aimed at streamlining the review and approval process for therapeutic products is seen as suitable for many NHPs. Products with potentially high risks will require the most comprehensive pre-market data submission and scrutiny as well as a higher level and frequency of post-market surveillance. Products of low risk will have minimal pre-market requirements and post-market assessments will be based on adverse event reporting. The Committee has borrowed from this PLF framework to develop one that is appropriate for NHPs.
One potential problem for NHPs lies in Health Canada's emphasis on particular types of scientific data in defining risk. When grouping products into the four broad categories (Category I to IV), risk is defined by the amount of knowledge/experience related to quality, efficacy and safety or known risk available on the product. On the highest risk level, Category IV products with little or no prior knowledge require comprehensive data and scrutiny while on the lowest risk level, Category I products with extensive knowledge require sponsor compliance with a pre-established monograph. Witnesses asserted that many NHPs, because of the current lack of knowledge or experience within Health Canada, would be grouped into the potentially high-risk category along with products for which there are identified safety issues.
Many witnesses readily acknowledged and supported the role of the federal regulatory system in evaluating products in terms of their benefits and risks. They also agreed that this evaluation must be based on adequate and accurate information. They did, however, have reservations about the appropriateness of using the current drug risk assessment model for NHPs. More specifically, they felt that assessments based on considerations of safety, quality, dosage, type of claim, seriousness of the disease and evidence of efficacy must be adapted for NHPs. They particularly argued that the model over-emphasized the risks of NHPs as a group and wanted more attention to the higher margin of safety associated with most products. In addition, they asserted that the requirements were slanted too strongly toward clinical trials and western science to the detriment of traditional knowledge and other culturally-based methods of assessment. They argued for a new approach that would, among other things, involve some changes to terminology, to conceptual underpinnings, and to evaluation processes.
For assessments of NHPs, the Committee favours a revised risk/benefit system that is more stringent than the one currently in place for foods but less stringent that the one applied to drugs. It agrees that the majority of NHPs are inherently safe and that regulatory efforts should be directed toward those that are less safe. In this regard, it notes that any product may be unsafe for reasons unrelated to its nature such as the way it is produced, stored or used. The following sections on safety, quality, efficacy and product licensing provide a more detailed discussion of how product evaluations might proceed for NHPs.
The vast majority of witnesses argued that most NHPs are safe and that assessments must be conducted with this view in the forefront. They emphasized that their uses are well known and pose minimal or no risk of harm. Witnesses noted that both mortality and morbidity rates associated with NHP use were negligible in comparison with pharmaceuticals. In fact, witnesses said that the majority of NHPs are safe if used correctly, that is when used for the appropriate indications and in correct doses. Some witnesses stated that, in the absence of scientific data to the contrary, a long history of human usage is generally sufficient evidence of a product's safety. They emphasized that it is not practical, necessary or economically feasible to conduct in vivo and clinical toxicological studies to establish the safety of most NHPs.
The Committee heard repeatedly that there were no reported deaths due to the consumption of vitamins, minerals, homeopathic preparations or traditional herbal remedies.
The discussions did point to the multiple elements that relate to safety and, in many instances, referred to factors that point in the opposite direction - toward danger. Thus, questions arose about the harm, toxicity, side effects, or risks associated with the use of a product. Witnesses acknowledged that assessing NHP safety meant testing for such things as acute and chronic toxicity of various dosage forms; evaluating finished products for contamination (bacteria, heavy metal, insect parts, artificial chemicals or pharmaceutical drugs); using epidemiological and toxicological data to identify short and long-term sensitivities and health outcomes. Others linked safety for consumers to standardization of potency, labelling, information, and professional practices.
The Committee heard from several international regulators about their method of assessing products with respect to safety. For example, Germany has a pharmacovigilance system for all medicinal products and has relied on this to ban certain products. In the United Kingdom (U.K.), the general trend is to upgrade control on products with demonstrated risk, for example, making them prescription only rather than banning them. For unlicensed herbal medicines, legislation lists products where there must be greater control over sale and supply. In Australia, restricted herbs must undergo a more extensive registration process than listed products.
Of particular concern was the fact that NHPs, like all forms of self-treatment, can present a potential risk to human health for several reasons. First, as some witnesses explained, the self-administration of any treatment may delay a patient from seeking qualified advice or cause a patient to abandon treatment without first seeking a professional opinion. They argued that people who do not receive appropriate treatment from the onset would eventually cost more to the system. Second, it was indicated that the scarcity of documented information on the interaction of NHPs with conventional medicines poses some problems with negative interactions. There is a need to identify herbal ingredients that may potentially interfere with specific categories of conventional drugs based on known phytochemical and pharmacological properties of certain herbs and documented side effects. Third, other witnesses asserted that some NHPs are more toxic and require precise controls on dosages. They asserted that products identified as presenting a lower margin of safety and higher risk should be available only through qualified practitioners. For example, with the exception of standard TCM formulas that have been used for a long period of time for minor conditions it was suggested that most TCM formulas should be done through a practitioner. In addition, some homeopathic practitioners argued that improper or prolonged use of preparations may be harmful.
The Committee believes that it is important that clear and complete information about complementary treatments be available to people who choose to self-medicate with these products. Cautions about negative interactions of NHPs with conventional medicines must be more readily available to health care professionals and the public. Research on these types of interactions needs to be conducted and disseminated as broadly as possible. These issues are addressed further in the following sections on labelling and informed choice.
On the particular question of the inherent safety of individual products, the Committee would like to stress the principles that guided its thinking. First and foremost, safety of NHPs is of primary concern. Members also agreed that the NHPs are different in nature from either food or pharmaceutical products. We accepted the contention of the many witnesses who asserted that the vast majority of NHPs are inherently safe and weighed this alongside the equally compelling argument that it is the regulator's duty to ensure that the products undergo some form of assessment for safety. We would like evidence to be drawn from a range of sources, historical and recent, traditional knowledge and contemporary science.
The level of regulation should be consistent with the level of safety associated with a particular product.
We contend that the current TPP assessment model is not appropriate for NHPs and that existing regulatory resources would be best directed toward those products that are less safe. Recognizing that there is no absolute assurance of safety under all circumstances, we nonetheless look to the regulator for a reasonable assurance of safety. As the later section on product licensing suggests, when products are identified as having a lower margin of safety and therefore a higher degree of risk, the final assessment should also involve expertise from outside the established regulatory system.
The Committee therefore recommends that:
The new regulatory authority assume primary responsibility for assessing safety of products;
General safety protocols be developed by the Expert Advisory Committee based on EAC judgements of reasonable evidence;
When necessary, this regulatory authority establish appropriate working groups to assess the safety of specific products.
C. Quality/Good Manufacturing Practices
Numerous questions from Committee members related to the quality of NHPs. They wanted to know how consumers could be certain about such factors as purity, potency, and cleanliness of the products they purchase. They heard repeatedly that Canadians must be assured that "what's on the label is in the bottle." In fact, quality was viewed as something to be scrutinized from product development through products for final sale. In that perspective, Good Manufacturing Practices (GMPs) appear to be of paramount importance. GMPs are internationally accepted standards governing the manufacturing and distribution process to ensure the quality of products. GMP standards apply to the premises, equipment, personnel, raw material (identification), finished product testing, sanitation/cleanliness and record keeping.
There are no GMP regulations for food products in Canada. Health Canada, however, has produced voluntary guidelines for the use of food manufacturers and inspection staff from the CFIA. With respect to drug products, the general regulatory framework governing GMPs is found in Division 2 of Part C of the Food and Drug Regulations. All drug establishments (manufacturing, wholesale, packaging, importing, distribution, testing) are expected to comply with GMP standards. Compliance is assessed by regular inspections of establishments. An Establishment License is delivered annually to establishments that comply with GMP regulations. Althoug NHPs sold as drug products must conform to these general GMP requirements, the TPP has developed supplementary GMP guidelines for herbal medicinal products and for homeopathic preparations (October 1996).
The TPP has also established a GMP Enforcement Directive (December 1997). This directive states that, although the TPP will work with establishments to help bring their operations into GMP compliance, it will not tolerate chronic non-compliance. In such situations, enforcement actions will be considered in order to prevent the distribution of potentially unsafe drug products. These actions may include among others: requests to voluntarily recall drug products sold, requests to voluntarily retain or dispose of drug products for sale, refusals at Customs for entry of drug products, and seizure of drug products for sale.
Like Canada, most developed countries require that drug products be manufactured pursuant to GMPs. In Australia, products are monitored before and after they are marketed, first through GMP inspections, and second through a sampling program (random testing) for lower safety products. Establishments must hold a license to manufacture therapeutic products. As in Canada, Australia has adapted its drug GMP guidelines to accommodate herbal products. In Germany, herbal products sold as drugs must be manufactured pursuant to GMPs. In the United Kingdom, all manufacturers of drug products must have a license and must comply with GMPs. As in Canada, they are inspected on a regular basis. In the United States, drug manufacturers must also satisfy GMP requirements. However, dietary supplements are not presently covered by drug GMP regulations. The legislation grants the Food and Drug Administration (FDA) the authority to establish GMPs specific to dietary supplements and the Committee was told that the FDA is in the process of establishing these regulations.
The vast majority of witnesses who discussed GMPs were of the view that manufacturing standards were necessary to ensure NHPs' quality and safety. The compliance to current GMP guidelines has resulted in extensive renovations and the purchasing of expensive equipment by Canadian establishments dealing with NHPs to ensure these high safety and quality standards. In fact, the Committee was told that Canadian GMPs are among the highest in the world and that Canadian NHPs gain international recognition because of their high quality.
However, most witnesses suggested that current GMPs were not appropriate for NHPs. When classified as drugs, NHPs must comply with standards initially developed for the manufacture of pharmaceutical products; these standards were said to be too stringent. By contrast, food standards may not be stringent enough for NHPs. The Committee was told that the inappropriateness of both food and drug GMP standards for NHPs has resulted in a lack of uniformity in GMP requirements across the country, because of unofficial concessions by field inspectors. For these reasons, witnesses recommended that all NHPs be manufactured according to appropriate GMPs. Specific GMP guidelines for NHPs would reflect their different nature, safeguard public safety and place no unnecessary burden on the NHP industry. Appropriate GMPs should be less costly than applying pharmaceutical GMPs and result in savings for both NHP industry and consumers. Witnesses also suggested that GMP standards address the specific needs of all types of products that make up the NHP group. Some of them, who noted that many herbal companies are small and medium-sized businesses, indicated that GMP standards should be flexible and adapted to the financial capabilities of these companies.
With respect to herbal products, botanical identity, purity and potency were stressed as particularly important factors in quality control. Poor quality of herbal products may be the result of substitution or contamination of the declared ingredients, with a more toxic botanical, a poisonous metal or a potent non herbal drug substance rather than of the pharmacological activity of the herbal ingredients themselves. The Committee was told that current GMP guidelines are inadequate because certification of botanical identity and purity testing are not mandatory and that criteria and methodology for determining identity are not specified.
The Committee agrees that NHPs sold on the Canadian market must meet high standards of safety and quality. Furthermore, the Committee acknowledges that GMP guidelines relevant to NHPs must be developed. These products are found in nature and it can be much more difficult to assess their quality and purity. Appropriate GMP guidelines will guard against packaged products that do not contain what they claim to contain. In order to ensure that Canadian products satisfy quality requirements, all manufacturers, packagers, importers and distributors selling NHPs to Canadians in Canada should hold a valid establishment license. This license would indicate continued compliance with GMPs. Overall, these observations are consistent with the Committee's principles regarding the nature of NHPs, quality and safety.
It is imperative to assure Canadian consumers that what is stated on the label is in the bottle
Therefore, the Committee recommends that:
Health Canada, in collaboration with the NHP industry, establish appropriate GMP guidelines reflective of the different nature of NHPs;
GMP standards for NHPs include specific quality control and testing for herbal products;
Manufacturers, packagers, importers and distributors of NHPs, whether located in Canada or abroad, be obliged to hold valid establishment licenses;
Inspection activities be performed consistently and on a regular basis by inspectors knowledgeable about the products.
The efficacy of a product is closely linked to the question of health claims. A product will only be efficacious if it produces the outcome indicated by its health claim. Thus, efficacy requires that a product "does what it says it will do."
Health claims, meanwhile, are statements of the effect of a product on the health of an individual made by the manufacturer or distributor, and displayed on the product label or literature. The Committee was told that there were generally three different categories of health claims. According to the APNHP, they are defined as follows. Structure-function claims report the effect of a product on a structure or physiological function in the human body and are based on the maintenance or promotion of good health. Risk-reduction claims relate consumption of a product to significant reduction in the risk of developing a disease or abnormal physiological state. Risk reduction may occur in two ways. One, the product may alter a recognized major health risk factor or factors of a disease or abnormal state. Two, it may affect a body function or system so as to improve the body's capacity to resist the disease or abnormal state. Therapeutic or treatment claims report the effects of a product on the actions of a specific disease or its symptoms. Treatment can include the cure or alleviation of either the disease or its symptoms.
EXAMPLES OF HEALTH CLAIMS |
|
| STRUCTURE FUNCTION CLAIM | "Calcium builds strong bones." |
| RISK REDUCTION CLAIM | "Garlic decreases the risk of cardiovascular diseases." |
| THERAPEUTIC OR TREATMENT CLAIM | "St. John's Wort is useful in the treatment of mild to moderate depression." |
| Source: Advisory Panel on Natural Health Products, Final Report, May 1998. |
The vast majority of witnesses contended that NHPs should be eligible to make all three types of claims if sufficient or reasonable evidence supports such claims. There was no unanimity, however, over what would constitute "sufficient" or "reasonable" evidence. For most witnesses, this could include clinical studies and/or traditional references and/or appropriate scientific data where available. Reliable traditional references could include those accepted in qualified jurisdictions - for example, the European Community, the United States or Australia. For some, professional consensus could also be a valid source of information about the use of NHPs. For others, sufficient or reasonable evidence means only scientific studies and controlled laboratory experiments.
The Committee was told that NHPs were different from pharmaceutical products and that the efficacy of natural products could not be assessed the same way as synthetic products. Many witnesses indicated that the efficacy of NHPs has been demonstrated over centuries through their use by a large number of people. Therefore, according to these witnesses, experience, clinical practice and empirical verification are evidence for NHPs' efficacy.
Some witnesses suggested that NHPs, particularly herbal products, be subject to the same rules required for other therapeutic products. Others felt that, in the absence of clinical trials, long-term records of use through peer-reviewed literature, along with a good risk assessment process, could be considered. Others stated that the current approval process needs major revisions to ensure that Canadians have access to products that are proven to be efficacious. They contended that clinical trials are necessary to prove efficacy and believe that clinical trials are not too expensive even for small companies. Products meeting requirements for safety and quality, but unable to demonstrate efficacy, could continue to be made available to Canadians but without health claims.
By contrast, the Committee was also told that it is the subjective satisfaction of consumers which is the critical measure. They should be the final arbiters. Others added that consumers and professionals would be the best judges of their own interests when they can freely weigh the evidence and opinions in the open market. As a result, they were of the view that efficacy should not be subject to government regulation.
Some witnesses indicated that the label should indicate the source for the therapeutic use, with phrases such as: "traditional use of this product indicates..." or "clinical use of this product indicates..." or "scientific studies have indicated..." Others recommended that products, for which no scientific evidence exists to support their use, attach a disclaimer such as: the effectiveness of this product is not supported by the usual scientific evidence required for non-prescription medications. It was also suggested that, if higher safety products were not to be subject to any pre-market approval, the label could include a disclaimer stating that the products have not been evaluated or approved by Health Canada for the diagnosis, treatment, cure or prevention of any disease.
Several witnesses indicated that the level of evidence needed to establish a health claim should be linked to the type of claim being made. Thus, treatment claims would need different evidence than structure/function claims. It was suggested that NHPs be permitted to make health claims only if two conditions were met: first, acceptable standards ensure that the active ingredients are present in meaningful amounts at the time of manufacture and throughout the shelf life of the product; and, second, the product is produced under conditions that meet acceptable GMP standards.
The Committee notes that there appears to be a trend in international regulation of NHPs toward more flexibility when it comes to assessing efficacy. For example, in Australia, efficacy is not perceived to be a priority for listed medicines. However, sponsors are required to keep efficacy data to be made available on request and claims are limited to minor self-limiting conditions. In the United States, dietary supplements are allowed to make health claims pursuant to the Dietary Supplements Health Education Act of 1994. Claims must be substantiated by the sponsor although pre-authorization is not required from the FDA. However, dietary supplements are not allowed to make drug claims.
The Committee agrees with the three categories of health claims as set out by the APNHP: structure-function claims, risk-reduction claims and therapeutic or treatment claims. The Committee believes that NHPs should be allowed to make all three types of claims if the rules as set out below are satisfied.
While the Committee agrees that consumers will be the final judges as to the effectiveness of a product, it does feel that the government has a role to play. If a person wishes to make a health claim about a product, we feel that reasonable evidence is required. This does not mean, however, that the evidence needed should be equivalent to that required for pharmaceutical products. No claims should be allowed where there is not appropriate evidence to support the claim. The Committee believes that this is the only way to ensure that consumers are making informed choices. If there is no basis for a claim, the product could still be sold without a claim as long as it satisfies the requirements regarding safety and quality. Adequate evidence to support health claims will help ensure that Canadians will be protected from fraudulent claims of health benefits.
The Committee rejects the suggestion that health claims should be allowed without some form of evidence.
We believe that the issue of health claims is related to the degree of safety or, conversely, the level of risk of the product. In fact, the dangers associated with product use can have as much to do with an unsubstantiated claim as with the toxicity of the product. If the claim is not valid, this can have severe consequences for the consumer. As noted earlier, the consumer might rely on self-treatment and delay seeking alternative advice or different treatment that could produce better health outcomes. The more important the claim, the higher the need to ensure that is based on accurate information and maximal safety for the consumer.
Thus, the Committee feels that the validity of a claim must be assessed. Because of the high safety of many of these products, pharmaceuticals standards are generally too rigorous. The Committee believes that the type of evidence needed should depend on the type of claim being made. For more serious claims, more rigorous evidence will be needed. While double blind clinical trials should be required for certain serious claims, other claims should require different evidence. Thus, unlike pharmaceuticals, the evidence that is required for certain NHP claims should be more flexible. They should include generally accepted and traditional references, professional consensus, clinical evidence including but not limited to double-blind trials and other types of clinical or scientific evidence. While the TPP currently accepts traditional references for traditional herbal medicines, the Committee believes that the system must be made more flexible to also recognize other types of evidence.
As described further in the section on product licensing, the Committee favours the development of monographs that provide pre-determined product information. Thus, products would have relevant monographs containing a standardized product description to which others must conform or by which others will be judged. The person wishing to make a claim would only have to attest that they satisfy the monograph. Where no such monograph exists or where the claim is not contained in a monograph, the person would have to provide some type of evidence. The following section on product licensing contains additional discussion about evidence. The type of evidence needed for structure-function claims and risk-reduction claims should be different from the type of evidence needed for treatment claims. In addition, the type of evidence needed for minor self-limiting diseases should differ from that for treatment of more severe conditions. For treatment claims for more severe conditions, double-blind clinical trials might be appropriate. The restriction imposed on health claims by Schedule A and its list of diseases and disorders is discussed in Chapter 7 of this Report.
The Committee concurs with witnesses who suggested that the label of the product should state clearly the nature of the claim (traditional use, clinical use, scientific studies). This will ensure that consumers know the basis for health claims thus allowing them to make informed choices. In addition, this should provide an incentive for the NHP industry to conduct further research with respect to NHP claims. Alternatively or in addition to the nature of the claim, the product could have attached a disclaimer that the effectiveness of this product is not supported by the usual scientific evidence required for non-prescription medications when such evidence was not presented. The Committee feels that this will provide a more level playing field between the pharmaceutical and NHP industries.
The Committee believes that this is consistent with its principle regarding safety since only products with reasonably proven effectiveness will be allowed to make claims. The procedure set out above also ensures that consumers will be allowed to make informed choices. This also keeps in mind the unique characteristics of NHPs and the need to respect cultural diversity.
Therefore, the Committee recommends that:
NHPs be allowed to make health claims, including structure-function claims, risk-reduction claims and treatment claims;
Claims be assessed to ensure that there is reasonable evidence supporting the claim;
The evidence not be limited to double blind clinical trials but also include other types of evidence such as generally accepted and traditional references, professional consensus, other types of clinical trials and other clinical or scientific evidence;
The evidence required vary depending on the type of claim being made, with different evidence being required for structure-function claims and risk-reduction claims for minor self-limiting conditions than for therapeutic or treatment claims;
The label indicates clearly the type of evidence used to support the claim.
Many witnesses appeared before the Committee to recommend the establishment of a new marketing process for NHPs. We received numerous suggestions regarding how products should be marketed. It is not practical to summarize all the recommendations that were presented to the Committee. However, we want all stakeholders to be assured that their recommendations were analyzed. The suggestions ranged from a notification system based on monographs, to a system based on the pharmaceutical model, to a system of total or partial deregulation.4
The Committee agrees that a new and more efficient product licensing system is needed for NHPs. While the Committee does not agree with post-market notification, as recommended by many witnesses, including the APNHP, it does see the need for a revised and more effective pre-approval process. As described in previous sections, the approval process must ensure that products are safe and that claims are supported by reasonable evidence. In addition, post-market surveillance (including GMPs) is also part of product licensing and helps to ensure the quality of products.
The Committee feels it is imperative that products be able to reach the Canadian market quickly when this is warranted.
The Committee agrees with many witnesses that the new process must be based on a risk management approach that recognizes that NHPs fall along a continuum of relative safety and therefore require different levels of control. The Committee was told that many NHP products are safe and therefore government intervention should be at a minimum. When a product is considered as having lower safety, the regulatory system should be more stringent. The system must keep in mind the higher margin of safety of the majority of NHPs. The Committee stresses, however, that in no circumstances should these products not be regulated. As stated earlier, the difference is that the level of regulation should be consistent with the level of safety associated with a particular product.
We are of the view that categories must be created within the NHP class to determine what level of regulation is appropriate for the particular products. For example, the APNHP suggested higher safety and lower safety classes. We do not feel qualified to determine what classes should be created but agree that products should be differentiated based on the risk they represent. This way of proceeding will ensure that the full impact of the regulation will only be felt by lower safety products. Health Canada should undertake the task of creating these classes in conjunction with the Expert Advisory Committee.
Obviously, the framework will not be an improvement if most NHP products are classified as lower safety products. The Committee does not believe this will occur since individuals with expertise and understanding of these products will now take decisions. Some of the factors used to determine higher or lower safety would surely include the inherent risks of the product, the type of claim being made, the seriousness of the disease, etc. For example, the safety of the product would only be one of the factors in determining the risk of the product. A product that is inherently safe could become a lower safety product if it makes a serious treatment claim. In addition, the way the product is produced could have an impact on its margin of safety. As was explained in previous sections, products that have lower safety margins would be held to different standards. In addition, the quality aspect would be assured with GMPs and Establishment Licensing. With respect to claims, evidence would vary depending on the claim.
The Committee acknowledges that the regulatory system set out in previous sections imposes stricter regulations as the risk of the product rises. The Committee believes this is the best approach for regulating NHPs. For higher safety products with minor claims, there would be different evidence needed than for a lower safety product making a treatment claim for a serious illness. The safety and effectiveness of a product should be based on submitted data and its assessment. The required data should also be consistent with the product's margin of safety.
As indicated earlier, the Committee favours a licensing system based on monographs containing previously agreed standardized product information against which other products could be assessed. The monographs could be developed from information already available in other countries as well as that provided by manufacturers and the Health Protection Branch. The information could then be reviewed and monographs issued as an authoritative standard for products entering the market. A major requirement for a person wishing to market the product would be to attest to the monograph. The product would then be able to make any claim referenced in the monograph, if all other conditions are satisfied. This would include structure function claims, risk reduction claims and treatment claims. As stated previously, we do not agree, however, that the notification to the regulator should be post-market. The person marketing the product should notify the regulator before the product is marketed. The regulator would have a short period of time (for example 30 days) to approve the application and issue an NHP number. Once the application is approved, the person would be allowed to market. This would allow the regulator to deal with any potential problems before the product is marketed.
The Committee notes that monographs for NHPs are not widely available in Canada. The Committee feels that Health Canada with designated working groups should review this issue and create new ones following acceptable formats and procedures. The Committee did hear that other countries have monographs and standards and therefore feels that these should be used as a basis for the creation of Canadian monographs. Obviously, these would have to be reviewed but we feel that there is no reason to replicate the work done in other countries. The creation of a Canadian pharmacopoeia where all monographs could be collected together would be a major undertaking. However, there is no reason that we could not start with individual standardized monographs consisting of information on product identity, claims, warnings, etc. with the long-term goal of creating a pharmacopoeia. Once more monographs achieve recognition in Canada, the NHP industry will be able to market products without unreasonable delay.
A product monograph is a document that describes the source, characteristics, biological activities, toxicity, pre- cautions, dosage and contraindications, etc., of a given plant or product.
With respect to products that do not have monographs, evidence should be presented to and reviewed by the regulator before they are marketed. Product assessment would be undertaken pursuant to the guidelines set out above and in previous sections. Once a product is approved, the regulator would issue an NHP number and provide what information is to be indicated on the label and other controls necessary to mitigate a product's potential harm.
The Committee also believes that post-market monitoring is crucial. The product should be easily tracked through its life cycle and its quality, safety and efficacy should be regularly monitored. As with the pre-market assessment, the Committee feels that the level of post-market monitoring should be based on the level of safety of the product. We are convinced that an NHP adverse event reporting system would be an important part of this post-market assessment. Therefore, establishments marketing products would be required to maintain and analyze post-market data of their products. For products that are of lower safety, more extensive reporting of adverse reactions would be required. Less frequent and detailed reporting would be needed for higher safety products. Reporting should be extensive enough to include interaction with other NHPs, with pharmaceutical products and with foods. The Committee also feels that an adverse reaction hotline must be made available to practitioners and the general public to report problems they may encounter with certain products. This hotline must be made as user friendly as possible. This would allow the regulator to better mitigate any risks associated with a product.
The Committee notes that Australia has an adverse reaction reporting system and this system is an important element of their post-market monitoring. Companies are obliged to report any known adverse reactions occurring during the marketing of a product. In addition, the Therapeutic Goods Administration (TGA, the equivalent of the TPP) encourages health practitioners to submit reports of adverse reactions and they have an Adverse Drug Reaction Committee that analyses reports and ranks them as probable, possible and not necessarily related to that particular substance. This helps in determining whether additional controls or warnings are needed or what action needs to be taken in relation to products. To date this has focused on conventional pharmaceuticals but is looking at ways to encourage practitioners of natural therapies to start reporting adverse reactions for these products.
The Committee desires a system that would recognize the different nature of NHPs while ensuring the safety and quality of accessible products. The overall result should be a system where lower safety products with a greater potential to cause harm would:
- require comprehensive pre-market submissions;
- undergo intensive scrutiny before being allowed on the market;
- sustain extensive controls designed to mitigate their risks;
- receive higher level and more frequent post-approval surveillance.
The Committee feels that this new framework allows the regulator to evaluate a product's potential risks (by assessing the safety of the product, by appraising the claims based on its seriousness and by ensuring compliance with GMPs) and potential benefits (by assessing product safety, efficacy and quality). Overall, it establishes a system to mitigate the risks when this is necessary (primarily by allowing additional information on labels and particular controls on the product). It is the view of the Committee that such a risk management approach is the best way to proceed.
Another aspect of risk mitigation is restrictions on the sale of certain lower safety products. The Committee feels that certain products should not be made available to the public before consultation with a qualified practitioner. The Committee realizes that it is the provinces that have the authority to regulate health-care practitioners and, in many cases, practitioners offering care that involves NHPs are not regulated. The Committee agrees that certain lower safety products should be made available pursuant to the approach suggested by the APNHP. Some lower safety products would be allowed for sale with cautionary labelling including the following:
- a statement that the product has been classified as low-safety by the regulatory body;
- a statement of the nature of the risks associated with the use of the product;
- a statement that the consumer should consult with a qualified practitioner before using the product.
The Committee would like to make it clear that certain lower safety products should not be made available even with these warnings. Practitioner intervention is an important aspect of risk mitigation although the Committee realizes that its implementation is problematic because few NHP practitioners are regulated. Thus, certain lower safety products should only be made available with practitioner intervention while other lower safety products would be made available with the statements listed above. Providing this information to consumers can mitigate the risks associated with a product.
The Committee notes that the proposed product licensing framework should lower the costs for the NHP industry. It sees two reasons for such an outcome: in many cases, less extensive data requirements will be required and reduced levels of regulation should be accompanied by lower fees.
In conclusion, the cornerstones of our new framework would be to identify the potential risks of NHP products and to control those risks where necessary. The risk of a product can be mitigated by product labelling and practitioner intervention and other controls on the product. Government intervention would be minimal unless clear safety concerns associated with a product emerge.
The Committee believes that the new framework should be phased in to allow sufficient time for the stakeholders and the regulator to modify the current DIN system and conform to the new regulations. A six-month phase in period would seem appropriate. The Committee believes this product licensing framework is consistent with our guiding principles with respect to the nature of NHPs, safety, quality, access, informed choice and costs.
Therefore, the Committee recommends that:
The new product licensing framework be based on a risk management approach that emphasizes the margin of safety associated with a particular product;
Health Canada, in conjunction with the Expert Advisory Committee, establish categories within the NHP class to determine what level of regulation is appropriate for a particular product;
A product licensing system based on monographs be used when they are available. Such a system should rely on a pre-market approval process and the regulator should have a short period of time (for example, 30 days) to review the application;
Health Canada, in conjunction with the Expert Advisory Committee, establish procedures to create new Canadian monographs based on work already accomplished in other countries;
Manufacturers of products that do not have monographs be required to provide evidence to Health Canada before a product is marketed. The level of evidence would be consistent with the margin of safety associated with the product;
The level of post-market monitoring be based on the margin of safety associated with the product and include an NHP adverse event reporting system for industry and an adverse reaction hotline for practitioners and the general public;
Certain lower safety products be made available to consumers with appropriate warnings and other lower safety products only be made available with practitioner intervention;
The new framework be phased in over a period of months to allow sufficient time for the stakeholders and the regulator to review the current DIN system and conform to the new regulations.
The Committee heard repeatedly that the current regulations severely limit the information that can be placed on NHP labels and that these restrictions do not allow consumers to make informed choices with respect to these products. Some even argued that current regulations are contributing to a vacuum of consumer information.
On the one hand, food labelling is very restrictive, as it does not allow information such as a product's potential benefits and how it should be used. On the other hand, drug labelling is also restricted, as NHP suppliers must conform to the use of information specified by Health Canada.
The Committee was told that there are risks arising from inadequate labelling. These risks include:
- over consumption by uninformed users;
- over dosage of children;
- usage in the presence of contraindicated conditions;
- failure to seek timely professional medical treatment;
- failure to recognize adverse effects (especially the more subtle chronic toxicity effects such as hepatotoxicity, teratogenicity and carcinogenicity);
- adverse herb-drug and herb-herb interactions;
- improper preparation and/or storage;
- improper application (ex. internal use of products intended for external application); and,
- allergic or adverse reactions due to undeclared ingredients.
Many witnesses requested that labelling be standardized. They provided a detailed list of the information that should be put on the label of NHPs. They were of the view that this type of information would allow consumers to make informed choices when selecting NHPs. The information requested included among others:
- health claims and/or therapeutic use (this subject has already been discussed previously);
- lot or batch number to allow for GMP quality control, and to facilitate recall action;
- expiration date to advise the consumer of the estimated shelf life of the product under normal storage conditions;
- special storage conditions if required;
- identification (scientific name) and amount (or proportion) of each ingredient;
- name and address of manufacturer;
- correct dosage for both adults and children and mode of administration;
- total number of dosage units (tablets, capsules, etc.) per package;
- quantity of ingredient(s) per dosage;
- warnings and contraindications for children, seniors, expectant and nursing mothers, people with specific medical conditions, and possible side effects;
- potential interactions with other NHPs or conventional medication or with foods.
Internationally, most developed countries have strict guidelines with respect to the labelling of therapeutic products. For example, Australia reviews labelling information during product assessment and warnings are permitted on labels. In the United Kingdom, a licenced product is required to have warnings while this is optional for unlicensed herbal remedies. In Germany, warnings and full labelling are required for drugs and warnings are even allowed on some foods. In the United States, if a product is a drug, a review of the labelling is part of the review process and there are strict guidelines with respect to labelling. Adequate directions and warnings must be included on the labelling. Food legislation also set out requirements for the labelling of foods. Internationally, the question of whether a product can make a claim generally revolves around the issue of whether the product is a food or a therapeutic product. Foods are generally not allowed to make health claims. One exception is the United States, where dietary supplements (foods) are allowed to make structure and function claims but not therapeutic claims.
The Committee agrees that consumers must be provided with all pertinent information when they are buying NHPs. Many of these products will be available over the counter and are intended for self-medication. In these circumstances, it is crucial that the consumer be permitted to make an informed choice regarding these products. This will also alleviate many of the potential risks associated with a certain product. Clearly, the information supplied on labels is linked to safety since it can be used to warn certain members of the population (i.e. pregnant women, the elderly, or children) that there is a danger for them in using a certain product. Also, if there is a potential hazard associated with the use of a product, particularly if it is being used outside its traditional setting, then this can be indicated on the label. In addition, the label could indicate to a consumer when consultation with a qualified practitioner is warranted or when health care providers should be informed of NHP product use. One of the benefits of detailed labelling is that it is a vehicle for education.
The Committee views extensive information on labels as a cornerstone of its risk management approach.
We wish to stress that pertinent information must be provided not only on the label that is actually on the product's container but also on other modes of information such as the packaging and inserts.
The Committee agrees with the recommendations that were made by the APNHP with respect to this issue: At a minimum, product information must encompass:
- identity of the product;
- directions for use;
- cautions, warnings or restrictions on the use of the product;
- lot batch number to allow for GMP controls and to facilitate recalls;
- expiry date to guide consumers on the expected shelf life under specified storage conditions;
- special storage conditions, if any;
- approved health claims.
The Committee feels that Health Canada should review this list in conjunction with the new Expert Advisory Committee to ensure that all relevant information will be available to consumers. We note that the following recommendations satisfy our principles of allowing consumers to make informed choices, of providing quality control measures and of enhancing the safe use of these products.
Therefore, the Committee recommends that:
Health Canada consult with its new separate NHP Expert Advisory Committee to determine what information is to be included on the labelling, consisting of, at a minimum, the items recommended by the Advisory Panel on Natural Health Products;
NHP labelling provide consumers with all relevant information needed to make informed choices;
NHP labelling be standardized to provide clear and consistent product information.
CHAPTER 7 - SECTION 3 AND SCHEDULE A OF THE FOOD AND DRUGS ACT
Section 3 and Schedule A of the Food and Drugs Act are inter-related. Subsections 3(1) and (2) prohibit advertising or selling to the general public of a food or drug as a treatment, preventative or cure of any of the diseases, disorders or abnormal physical states referred to in Schedule A. Section 3 states the following:
|
1) No person shall advertise any food, drug, cosmetic or device to the
general public as a treatment, 2) No person shall sell any food, drug, cosmetic or device (a) that is represented by label, or (b) that the person advertises to the general public as a treatment,
preventative or cure for any of the |
Some of the diseases, disorders or abnormal physical states listed in Schedule A include alcoholism, arthritis, asthma, cancer, depression, diabetes, disease of the prostate, heart disease, liver disease and many more. Diseases can be added or deleted from the Schedule by regulation. The effect of these provisions is an outright prohibition even though there might be evidence supporting a health claim.
Apparently, the purpose of these provisions and Schedule A is to ensure that individuals will seek medical attention for serious diseases, to restrict advertising when self-diagnosis and self-treatment is not considered advisable and to limit the possibility of fraudulent claims being made with respect to foods and drugs.
The Committee was told that section 3 in combination with Schedule A can unintentionally restrict the dissemination of information that can be beneficial to consumers and be in the interests of public health. Apparently, the impact is more severe on products from other cultures, particularly traditional Chinese products, many of which are used for conditions listed in Schedule A. Witnesses added that the Schedule is outdated and no longer reflects the reality of products available on the market today. The Committee was told that the Schedule should be eliminated or at the very least made more flexible. Other witnesses did not recommend that Schedule A be eliminated but that the list of diseases be reexamined. In the short term, section 30 should be invoked to remove diseases currently listed in Schedule A.
The Committee notes that some of the objectives of section 3 and Schedule A appear to be satisfied by other regulatory measures. For example, in cases where self-diagnosis and self-treatment is not considered advisable, the product can be made available by prescription only. This restricts product availability to individuals who have obtained advice from a health practitioner. This also restricts the advertising of such a product to the public since prescription products cannot make representation other than with respect to the brand name, proper name, common name, price and quantity of the drug. Thus, advertising is limited even without recourse to section 3 and Schedule A. For non-prescription drugs, it is not clear why those dealing with Schedule A diseases should be prohibited. Health Canada would have assessed the safety and efficacy of the product and any unwarranted claim would not be approved.
With respect to the prevention of fraud, it should be remembered that foods are generally not allowed to make health-related claims. With respect to drug claims, as stated above, Health Canada must first approve them. In addition, sections 5 and 9 prohibit advertising that is false, misleading or deceptive with respect to both foods and drugs. Thus, it is not clear that Schedule A is needed to prohibit fraudulent claims since other provisions of the Act and its Regulations already seem to regulate such activities.
The Committee is aware that similar limitations are applied in other countries. In the U.K. there are fairly stringent regulations on the types of claims allowed for medicines. Generally, they are limited to minor self-limiting diseases and there are regulations listing certain conditions for which advertising is not permitted, for example, bone and cardiovascular diseases. Germany also has a catalogue of diseases for which products are not allowed to be advertised for purchase by the general public. An expert committee continually updates this list. In Australia, claims for minor self-limiting conditions of listable products are related to the Therapeutic Goods Advertising Code which sets out the guidelines for what is permitted in public advertising. The Code is developed in a co-regulatory approach through a committee, the Therapeutic Goods Advertising Code Council, which comprises of industry, professionals, consumers and regulatory representatives. It should be noted that the Code sets out a negative list of indications, disease states that may not be referred to in advertising or claims, for example, heart disease, diabetes and cancer. There is also a complaint mechanism for consumers who feel that sponsors have exceeded permitted claims. The Council is currently reviewing the advertising Code.
The Committee feels that the current provisions may unduly restrict health promotional advertisement that may be beneficial to consumers and may prevent self-medication in cases where it is warranted. At a minimum, the diseases included in Schedule A should be thoroughly reviewed to ensure that only appropriate diseases are included in the list. In addition, we observe that many of the diseases listed in Schedule A are broadly defined. Thus, we feel that, in appropriate cases, specific diseases should be exempted by regulation from the broadly defined term and consequently from the requirements of subsections 3(1) and (2).
The Committee also believes that Health Canada should conduct a study to determine whether Schedule A still serves a purpose and whether either subsections 3(1) and (2) should be deleted or all of the diseases be removed from Schedule A. The study should be conducted with participation from stakeholders representing consumers, the food, natural health product and pharmaceutical industries, and health practitioners.
These changes appear necessary considering the new products that should become available in Canada as a result of the new regulatory framework for NHPs. The implementation of these strategies would be consistent with our principle of providing more information to consumers and at the same time keeping in mind safety concerns; it would allow consumers to make informed choices while still controlling fraudulent advertising. This is also compatible with our principle of respecting diverse cultural traditions.
Therefore, the Committee recommends that:
Health Canada immediately initiate a review of the diseases listed in Schedule A to ensure that only appropriate diseases are included and, where relevant, specific diseases be exempted by regulation from the broad terms found in Schedule A;
Health Canada, subsequently, conduct a study with the participation of representatives from consumer groups, the food, natural health products and pharmaceutical industries, and health practitioners to determine whether subsections 3(1) and (2) of the Food and Drugs Act or all of the diseases listed in Schedule A should be deleted.
4 Only a limited number of witnesses discussed deregulation. Total deregulation means that no regulations whatsoever (with respect to pre-market approval or GMP requirements or labelling standards, etc.) would govern the sale of NHPs on the market. Under such a system, the role of government is very minimal and limited to proving that a product may be or has been harmful. Partial deregulation refers to a relaxation of some of the current regulations.